


La cheratopatia verticillata rappresenta una condizione non inficiante la vista che si caratterizza per la presenza di depositi corneali intra-epiteliali di particelle lipidiche di colore giallo-bruno che, a partire da un punto paracentrale inferiore della cornea, si distribuiscono con andamento a vortice fino alla periferia corneale. Sono sempre bilaterali e sebbene solitamente non compromettano l’acuità visiva, il paziente può talora lamentare offuscamento visivo, abbagliamento, fotofobia e aloni intorno alle luci.
Le possibili cause di cornea verticillata comprendono le seguenti qui elencate.
- L’esposizione a polvere di silicio tipica di alcuni settori professionali tra cui:
- industria estrattiva e lavorazione di materiali lapidei (es. rocce impiegate come materiali da costruzione o pietre ornamentali per rivestimenti di pregio);
- produzione di ceramiche (sotto forma di quarzo ventilato, presente in impasti e smalti vetrosi);
- produzione del vetro (la sabbia silicea è una delle materie prime utilizzate)
- cementifici (presente nella miscela usata per la produzione del clinker, componente base per la produzione del cemento);
- fonderie (la sabbia silicea viene usata per gli stampi in cui viene versato il metallo fuso);
- settore edile (ad esempio per malte, cemento, laterizi, rivestimenti in pietra naturale o sintetica, ecc);
- produzione di laterizi;
- settore farmaceutico (ad esempio per la produzione di paste dentarie);
- settore tessile (nelle operazioni di sabbiatura di tessuto denim).
-
Le terapie di lungo corso con alcuni farmaci e in particolare:
- Amiodarone (antiaritmico: FA e tachicardie ventricolari ricorrenti)
- Tamoxifene (antitumorale: modulatore selettivo dei recettori degli estrogeni)
- Naprossene (antiinfiammatorio non steroideo)
- Idrossiclorochina (antimalarico e antireumatico: si utilizza spesso per il trattamento di artrite reumatoide e lupus eritematoso sistemico)
- Indometacina (antiinfiammatorio non steroideo con forte potere analgesico)
- Iniezione sottocongiuntivale di gentamicina
- Il mieloma multiplo, una neoplasia maligna tipica dell’età avanzata (nell’80% dei casi colpisce pazienti over 60 anni) che colpisce le plasmacellule, cellule molto importanti del sistema immunitario, originate nel midollo osseo. Derivano dai linfociti B che, insieme ai linfociti T, sono fra le principali cellule coinvolte nella risposta immunitaria. Compito delle plasmacellule è produrre e liberare anticorpi per combattere le infezioni. Se la loro crescita diventa incontrollata possono dare origine al tumore. Le cellule di mieloma producono in grande quantità una proteina nota come componente monoclonale (componente M), che è un particolare tipo di anticorpo. Producono anche una sostanza che stimola gli osteoclasti, responsabili della demolizione del tessuto osseo e, di conseguenza, i pazienti colpiti da mieloma sono spesso soggetti a fratture ossee. Frequenti manifestazioni comprendono, oltre al dolore osseo, insufficienza renale, ipercalcemia, anemia e infezioni ricorrenti. La diagnosi richiede in genere la dimostrazione della proteina M (talvolta presente nell'urina e non nel siero e raramente assente totalmente) e di lesioni osteolitiche, di una proteinuria a catene leggere o di un eccesso di plasmacellule nel midollo osseo per cui è in genere necessario eseguire una biopsia del midollo.
- La malattia di Fabry, una delle più comuni malattie da accumulo lisosomiale. Malattia ereditaria legata al cromosoma X, caratterizzata da difetto del catabolismo dei glicosfingolipidi per una attività deficitaria dell’ enzima lisosomiale a-galattosidasi A (a-Gal-A). Il deficit enzimatico causa un progressivo accumulo di globotriaosilceramide (Gb3) e glicosfingolipidi correlati nei lisosomi delle cellule endoteliali e, in minor grado, delle cellule epiteliali, periteliali e cellule muscolari lisce.
Caso clinico
Paziente donna di 36 anni giunge alla nostra attenzione lamentando visione confusa in entrambi gli occhi da diversi anni. In anamnesi riportava buono stato di salute generale, una allergia all’acido acetilsalicilico e una storia di cheratite di ndd già sottoposta a numerosi controlli nei 4 anni precedenti alla nostra osservazione.
La paziente era miope, portatrice abituale di lenti a contatto, la cui acuità visiva al meglio della correzione ottica risultava essere di 12/10 in entrambi gli occhi.
Alla biomicroscopia alla lampada a fessura si poteva osservare bilateralmente la presenza di una importante cheratopatia verticillata (figure 1 e 2) con estensione dei depositi fino al limbus (figura 3).


In virtù dell'evidenza clinica si è deciso di procedere a una più accurata raccolta anamnestica che ha escluso l’esposizione ad agenti tossici (nello specifico la polvere di silice) e l’assunzione di farmaci quali in particolare tamoxifene, indomatacina e amiodarone. La paziente, a domande mirate, riferiva inoltre dolori aspecifici riportati in anamnesi fin dall’età adolescenziale, talvolta associati a parestesie e disestesie coinvolgenti le estremità.
Durante la visita la paziente era stata accompagnata dalla madre che non lamentava disturbi visivi ma che riferiva una storia positiva per un riscontro di cardiomiopatia ischemica apicale all’età di 40 anni mai trattata con antiaritmici, associata ad ipoacusia sinistra. Alla valutazione alla lampada a fessura anche la madre presentava un quadro di cheratopatia verticillata bilaterale (figura 4).
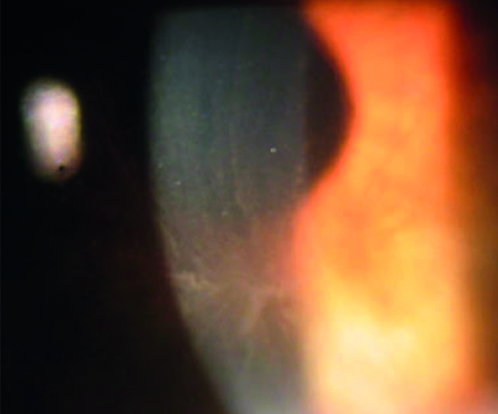
Abbiamo quindi posto sospetto diagnostico di malattia di Fabry per cui entrambe le pazienti sono state indirizzate all’indagine genetica presso uno dei centri di riferimento in Regione Lombardia, attraverso il quale si è evidenziata positività per una mutazione del gene GLA presente in entrambe le pazienti, con associato patologico aumento del marcatore biochimico liso-Gb3. Tale riscontro ha posto inoltre indicazione ad eseguire indagine familiare tutt’ora in corso.
Discussione
La malattia di Anderson-Fabry o, più semplicemente malattia di Fabry, prende il nome dai due dermatologi che per primi la descrissero, in modo indipendente, nel 1898: William Anderson, in Inghilterra, e Johannes Fabry in Germania.
È una condizione rara che può colpire maschi e femmine di tutte le età e di qualsiasi etnia. Si tratta di una condizione genetica legata alla inattivazione del cromosoma X e definita come una malattia da accumulo lisosomiale progressiva. Uno degli enzimi normalmente prodotti dall’organismo è chiamato enzima alfa-galattosidasi A o, per brevità, alfa-GAL. L’alfa-GAL provvede alla degradazione di una sostanza grassa denominata globotriaosilceramide, anche detta GL-3 o Gb3. Nelle persone con malattia di Fabry, l’organismo non produce una quantità sufficiente di alfa-GAL, oppure l’enzima è presente ma non funziona correttamente. Ciò significa che la globotriaosilceramide (GL-3) non può essere degradata normalmente e finisce quindi per accumularsi nei lisosomi compromettendone la funzione di “scavanger”.
Il gene che produce l’enzima alfa-galattosidasi, detto alfa-GAL, è situato sul cromosoma X per cui la malattia di Fabry si esprime in modo diverso negli uomini e nelle donne.
Gli uomini con malattia di Fabry avranno sempre un gene alfa-GAL difettoso, in quanto possiedono un solo cromosoma X. Ciò significa che gli uomini con malattia di Fabry generalmente non dispongono di alcun enzima alfa-GAL attivo o solo di una quantità molto piccola.
Le donne possiedono due cromosomi X e, se affette da malattia di Fabry, avranno in genere un cromosoma con il gene difettoso e un cromosoma con il gene correttamente funzionante. Nelle cellule di un organismo femminile, uno dei cromosomi X viene inattivato nel contesto di un processo chiamato inattivazione del cromosoma X (indicato anche come lyonizzazione). Si tratta di un processo casuale che avviene in tutte le cellule e che quindi potrà condurre a tre diverse condizioni:
- nella maggior parte delle cellule viene inattivato il cromosoma X funzionante;
- nella maggior parte delle cellule viene inattivato il cromosoma X difettoso;
- in un numero all'incirca uguale di cellule viene inattivato il cromosoma X funzionante o il cromosoma X non funzionante.
Come conseguenza di tale variabilità, i sintomi, la loro gravità e l’età a cui insorgono possono variare significativamente nelle donne, pur potendo essere altrettanto gravi che negli uomini.
I sintomi comprendono:
- dolore definito come urente o puntorio alle mani e ai piedi e che si irradia a tutto il corpo
- sudorazione ridotta o assente
- intolleranza al caldo/freddo
- intolleranza allo sforzo fisico
- eruzioni cutanee (angiocheratomi)
- cheratopatia verticillata e/o teleangectasie retiniche
- disturbi dell’udito
- diarrea, vomito o dolore addominale
- problemi cardiaci tra cui alterazioni della conduzione, ipertrofia ventricolare sinistra, aritmia e ischemia
- problemi renali con albuminuria, proteinuria e insufficienza renale progressiva
- problemi a carico del sistema nervoso prevalentemente di tipo ischemico (TIA, ictus)
- problemi neuropsichiatrici e depressione.
Bisogna precisare che non esiste alcuna cura per la malattia di Fabry, ma, una volta effettuata la diagnosi, sono disponibili trattamenti per controllare i sintomi e la progressione della patologia. In molti paesi, compresa l’Italia, sono inoltre disponibili terapie enzimatiche sostitutive (enzyme replacement therapies, ERT) o “chaperone farmacologici” (PCT). Le ERT si propongono di sostituire l’enzima naturale e di permettere così la rimozione di GL-3 dalle cellule, impedendone l’ulteriore accumulo lisosomiale; da qui l’importanza di una diagnosi certa e tempestiva che, come nel nostro caso, talvolta può passare in prima istanza per l’oculista.
Il focus sulla cornea prosegue domani, resta aggiornato! Domani "Infezioni corneali: criteri per una diagnosi eziologica".
Bibliografia
-
Francois, “Cornea verticillata,” Documenta Ophthalmologica, vol. 27, no. 1, pp. 235–250, 1969.
-
François, M. H. Hanssens, and H. Teuchy, “Corneal ultrastructural changes in Fabry’s disease,” Ophthalmologica, vol. 176, no. 6, pp. 313–330, 1978.
-
Ciancaglini, P. Carpineto, E. Zuppardi, M. Nubile, E. Doronzo, and L. Mastropasqua, “In vivo confocal microscopy of patients with amiodarone-induced keratopathy,” Cornea, vol. 20, no. 4, pp. 368–373, 2001.
-
Ikegawa Y, Shiraishi A, Hayashi Y, Ogimoto A, Ohashi Y, “In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease”, J Ophthalmol. 2018 Mar 21;2018:5315137.
-
Germain DP. Fabry disease. Orphanet J Rare Dis 2010, 5:30.
-
https://www.malattiadifabry.it
-
Desnick R.J., Ioannou Y.A., Eng C.M. (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet A.L., Vogelstein B, Kinzler K.W., Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds)





