


INTRODUZIONE
La Sindrome irido-corneo-endoteliale (ICE) rappresenta uno spettro di disordini monolaterali dell’endotelio corneale, dell’angolo camerulare e dell’iride spesso associati a disturbi visivi lievi o addirittura non associati a deficit funzionali,
sebbene talvolta sia possibile un calo visivo secondario al glaucoma o si possa verificare nel tempo una progressione verso un edema corneale invalidante.
La Sindrome ICE comprende diverse varianti tra cui:
- l’atrofia iridea progressiva;
- la sindrome di Cogan Reese o del nevo irideo;
- la sindrome di Chandler.
Le caratteristiche cliniche includono sinechie periferiche anteriori, anomalie iridee (corectopia, noduli e fori iridei), glaucoma, edema corneale e un caratteristico aspetto ad argento martellato dell’ endotelio corneale (T.C. Lucas-Glass, 1997).
Tra gli aspetti tipici, alla microscopia speculare c’è l’arrotondamento dei margini cellulari, un aumentato pleomorfismo cellulare e la perdita del mosaico cellulare.
Nel 1978 Campbell e i suoi collaboratori denominarono tale quadro “degenerazione proliferativa endoteliale primaria” a voler sottolineare come le alterazioni iridee e il glaucoma fossero in realtà secondarie ad una anomaliadell’endotelio corneale. Il comune legame tra le diverse varianti della sindrome ICE è proprio nella capacità di cellule anomale dell’endotelio corneale di proliferare e migrare attraverso l’angolo verso la superficie iridea. La membrana neo formata così generatasi, proliferando, ricopre l’angolo camerulare, si riflette sull’iride e successivamente si contrae determinando il glaucoma da chiusura d’angolo e le trazioni iridee che possono esitare in fori a tutto spessore.
Numerose evidenze scientifiche (Qi-Hua Le, 2009) hanno dimostrato che le cosiddette “ICE cells” sono responsabili dell’insorgenza della malattia e sono presenti in tutti i pazienti affetti. Si tratta di cellule dell’endotelio corneale anomale, trasformate nel senso di una epitelizzazione, con microvilli, filopodia e vescicole sulla superficie cellulare, espressione di citocheratina e vimentina nel citoplasma, e una tendenza alla replicazione in strati endoteliali multipli. Queste cellule possono ricoprire l’endotelio per intero o essere presenti solo in aree circoscritte (D.C. Garibaldi,
2005).
Tutto ciò spiegherebbe la comparsa tardiva dei sintomi anche se le cause precise della sindrome rimangono ancora sconosciute; tipicamente la sindrome ICE si manifesta in giovani adulti, solitamente di sesso femminile ed è quasi sempre monolaterale ma, ad oggi, ancora è ignoto quale ne sia il meccanismo patogenetico alla base.
Tra le ipotesi eziologiche proposte abbiamo infezioni perinatali o postnatali precoci, anomalie della cresta neurale, patologie neoplastiche e infezioni virali come quelle da Epstein Barr o Herpes Simplex.
In taluni casi il primo sintomo è la riduzione visiva nell’occhio coinvolto, tipicamente presente al mattino, quando l’edema corneale è più grave. La disidratazione del tessuto corneale, a contatto con l’aria nell’arco della giornata, comporta poi progressivamente una tendenza al miglioramento. Nei casi più gravi il paziente può soffrire di una riduzione cronica del visus dovuta all’edema corneale o all’atrofia ottica glaucomatosa. Il dolore è poi generalmente causato dall’edema e solo raramente dall’aumento del tono oculare (V. Orfeo, 2005).
CASO CLINICO
Nell’aprile 2005 è giunta per la prima volta alla nostra attenzione una donna di 39 anni con anamnesi oculare sostanzialmente muta (vale a dire negativa per dolore o traumi oculari pregressi), lamentando un calo visivo associato alla comparsa di visione offuscata al mattino nell’occhio destro. Dalla documentazione in possesso della paziente si evidenziava alle pregresse valutazioni oftalmologiche una lieve corectopia con la pupilla destra stirata lungo l’asse verticale; a causa del calo visivo progressivo e della persistenza della corectopia, la paziente era inoltre stata sottoposta a valutazione neurologica e a RMN encefalo, entrambe risultate negative. A quel punto la paziente era appunto stata inviata presso un ambulatorio di Patologia Corneale per un’ulteriore valutazione.
Il nostro esame obiettivo evidenziava un’acuità visiva pari a 6/10 con 0,50 sf // -0,50 cyl ax 170° nell’occhio destro e pari a 10/10
con -0,50 sf nell’occhio sinistro. La pressione intraoculare (IOP) risultava essere pari a 23 A mmHg in OD e 14 A mmHg in OS con uno spessore corneale centrale pari rispettivamente a 592 um e 515 um al pachimetro a ultrasuoni. La paziente riferiva di instillare da 6 mesi Dorzolamide collirio tre volte al dì.
L’esame alla lampada a fessura evidenziava una pupilla stirata lungo l’asse verticale con riflessi fotomotori alterati (figg. 1 e 2),
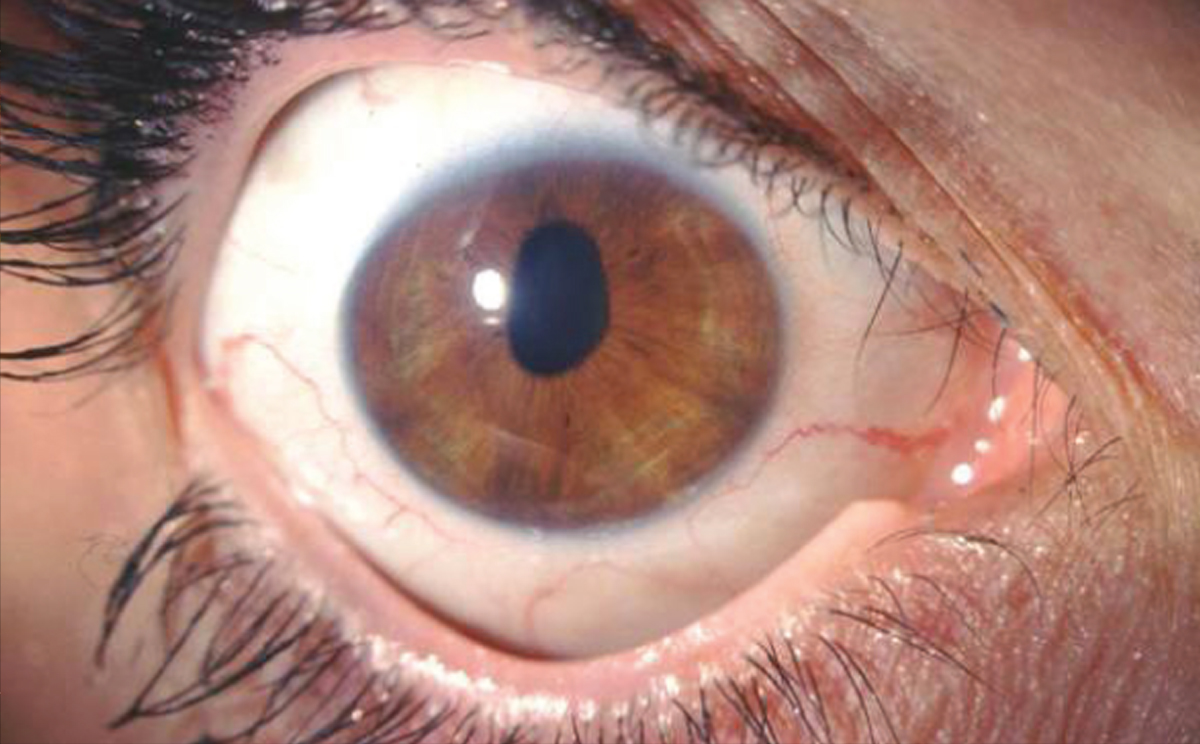
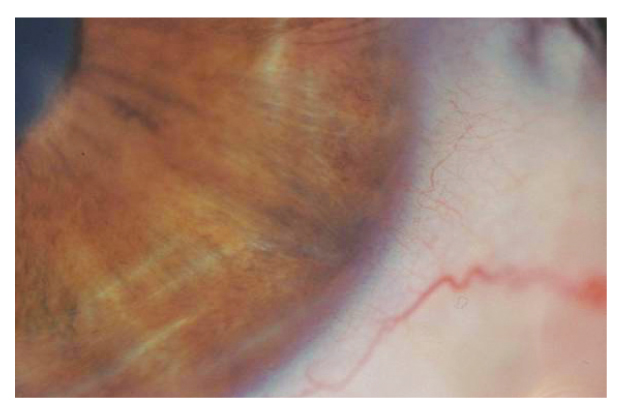
un modesto edema corneale con fini bolle epiteliali e un aspetto dell’endotelio corneale ad argento martellato (figg. 3 e 4).
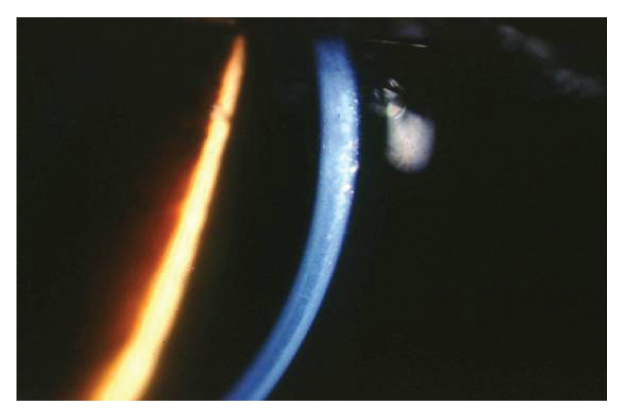
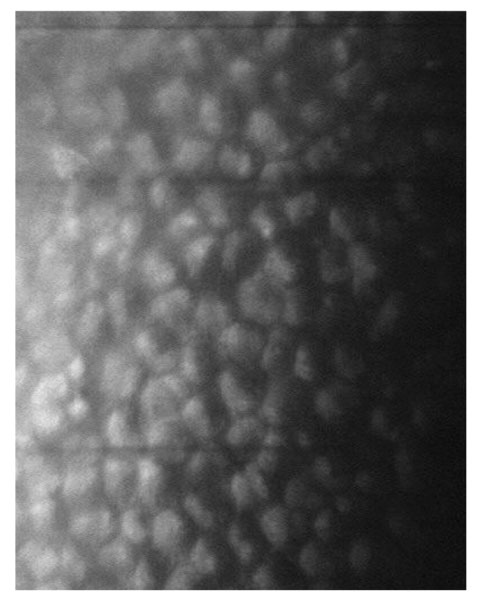
La biomicroscopia speculare non a contatto mostrava inoltre cellule endoteliali ingrandite, a margini arrotondati e iperriflettenti con citoplasma scuro e punto centrale brillante corrispondente alla protrusione del nucleo al di sopra di un citoplasma rarefatto (fig. 5).

L’esame con lampada a fessura dell’occhio sinistro non evidenziava invece alcuna anomalia ed anche l’ispezione del fundus oculi in dilatazione in entrambi gli occhi risultava essere nei limiti di norma.
Nonostante il controllo della IOP con la Dorzolamide il calo visivo e l’annebbiamento mattutino risultavano essere persistenti per via del persistere dell’edema corneale.
Ecco perché abbiamo deciso di trattare l’ipertono con un’associazione di timololo e latanoprost.
La paziente è stata quindi seguita negli anni onde valutare la progressione della patologia e al nostro ultimo esame (settembre 2014) i reperti nell’occhio destro risultavano essere sostanzialmente invariati eccezion fatta per la IOP (16 A mmHg) e lo spessore centro corneale sceso da 592 um a 547 um.
DISCUSSIONE
La sindrome ICE è una condizione rara e spesso erroneamente non diagnosticata che include una serie di quadri patologici tra cui l’atrofia iridea essenziale progressiva, la sindrome di Chandler e la sindrome di Coogan Reese. L’osservazione attenta dell’endotelio è fondamentale per la diagnosi e per la differenziazione di questa forma da altre rare endoteliopatie.
Le teorie eziopatogeniche alla base di questo disturbo includono (JA Alvarado, 1986):
- la teoria della membrana che suggerisce che un’anomalia dell’endotelio corneale porterebbe all’edema corneale e all’estensione di una membrana simil Descemet che incrocerebbe e ricoprirebbe l’angolo camerulare;
- le infezioni virali che potrebbero alterare il genoma cellulare conferendo capacità proliferative all’endotelio corneale;
- fattori esogeni e patologie traumatiche.
Le più comuni condizioni da porre in diagnosi differenziale con la sindrome ICE sono riassunte nella tabella.
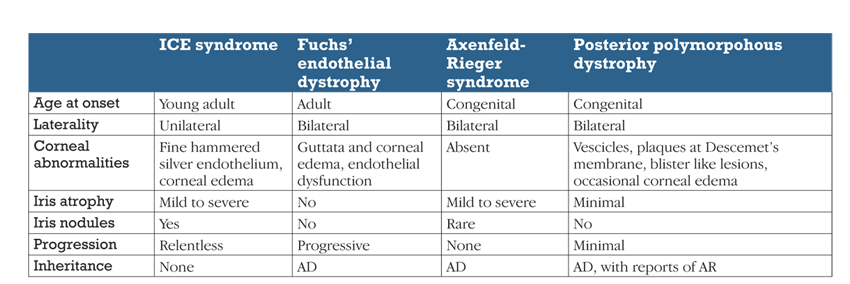
La nostra paziente ha presentato le tipiche caratteristiche della cosiddetta sindrome di Chandler con edema corneale diffuso derivante da una disfunzione endoteliale.
Non a caso la paziente riferiva un peggioramento della sintomatologia visiva al mattino con un miglioramento nell’arco della giornata grazie all’incremento dell’evaporazione in aria ambiente e all’aumento della pO2.
Inoltre nel nostro caso abbiamo anche osservato quella che è l’influenza della Dorzolamide su un endotelio funzionalmente compromesso (K. Inoue, 2003). La Dorzolamide è infatti un inibitore della anidrasi carbonica e sopprime la produzione di acqueo inibendo lo step anidrasi carbonica dipendente della secrezione di questo da parte del corpo ciliare; il tutto si traduce in una riduzione della IOP senza gli effetti collaterali degli inibitori dell’anidrasi carbonica sistemici (Acetazolamide).
Accanto ai processi ciliari, anche l’endotelio corneale possiede degli isoenzimi dell’anidrasi carbonica I e II. Essi giocano un ruolo nella funzione di pompa che regola il flusso di acqua al di fuori dello stroma corneale mantenendone la trasparenza. Ecco perché la Dorzolamide topica può influenzare il contenuto corneale di acqua e lo spessore corneale in endoteli funzionalmente compromessi, pur mantenedo la sua efficacia terapeutica ipotonizzante.
CONCLUSIONI
Abbiamo quindi presentato un caso di sindrome di ICE discutendo di quale sia in questi casi il trattamento ipotonizzante più efficace nella nostra paziente che ad oggi, dopo 9 anni di follow-up, presenta ancora condizioni cliniche piuttosto stabili. Il controllo della IOP in questi pazienti, infatti, può essere molto difficoltoso e spesso è necessario intervenire chirurgicamente.
Il solo approccio terapeutico possibile in questi casi è il trattamento dei sintomi e delle sequele poiché non abbiamo modo di arrestare la proliferazione endoteliale né la formazione di sinechie anteriori una volta che queste inizino a formarsi. Ecco perché il glaucoma e l’edema corneale sono a tutt’oggi i problemi clinici principali sebbene siano necessari ulteriori approfondimenti in merito all’agente causale della patologia.
BIBLIOGRAFIA
- Konowal, JM. Irreversible corneal decompensation in patients treated with topical dorzolamide. Am J Ophthalmol 1999; 127: 403-6.
- DC Garibaldi, OS. Features of the iridocorneal endothelial syndrome on confocal microscopy.
- Cornea 2005; 24: 349-51.
- JA Alvarado, CM. Pathogenesis of Chandler’s syndrome, essential iris atrophy and the Coogan Reese syndrome. Invest Ophthalmol Vis Sci 1986; 27: 853-72.
- K Inoue, KO. Influence of Dorzolamide on corneal endothelium. Jpn J Ophthalmol 2003; 47: 129-33.
- Qi-Hua Le, XH SJ. In-vivo confocal microscopy of iridocorneal endothelial syndrome. Int Ophtalmol 2009; 29: 11-8.
- Eagle RC, SJ. Iridocorneal Endothelial Syndrome with contralateral Guttate Endothelial Dystrophy. Ophthalmology 1987; 862-70.
- Laganowski HC, SE. Distinguishing features of the iridocorneal endothelial syndrome and posterior polymorphous dystrophy: value of endothelial specular microscopy. British Journal of ophthalmology 1991; 212-6.
- Lee WE, MG. Corneal Endothelial Cell abnormalities in an early stage of the iridocorneal endothelial syndrome. British Journal of Ophthalmology 1994; 624-31.
- Orfeo V, PM. ICE sindrome(Sn. Irideocorneoendoteliale): una nostra esperienza. Viscochirurgia 2005; 59-66.
- TC Lucas-Glass, KH. The contralateral corneal endothelium in the ICE syndrome. Arch Ophtalmol 1997; 115:40-4.





