


Anamnesi e indagini
Giunge alla nostra osservazione una giovane donna di 28 anni lamentando da circa tre giorni calo della vista bilaterale associato a cefalea, dolore orbitario ai movimenti oculari e saltuari episodi di acufeni e vertigini. La paziente gode di buona salute generale e non riferisce comorbidità a livello sistemico, né a livello oculare. All’esame obiettivo si riscontra un visus di 10/10 con correzione in entrambi gli occhi, la camera anteriore è in quiete e il tono è nei limiti di norma.


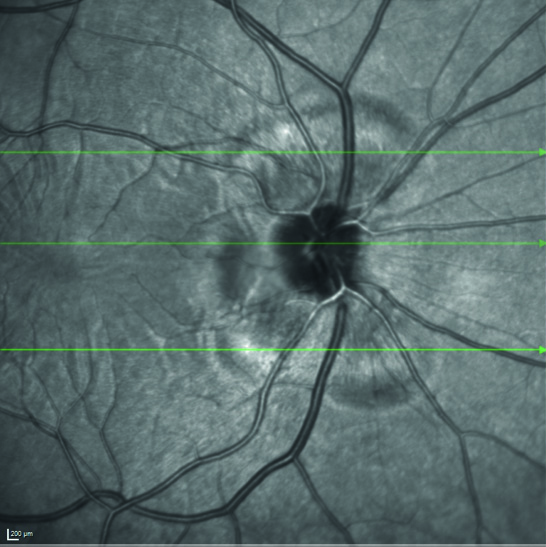
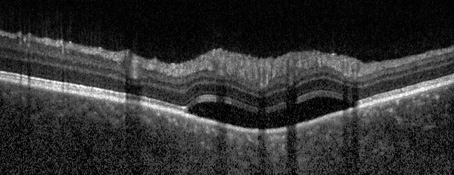
Esaminando il fondo oculare si rileva una lieve iperemia del nervo ottico maggiormente evidente in occhio sinistro rispetto al destro e la presenza di rare cellule vitreali. Eseguiamo quindi una tomografia a coerenza ottica (OCT) riscontrando al polo posteriore in entrambi gli occhi un diffuso ispessimento coroideale associato a sinistra ad un piccolo distacco del neuroepitelio temporalmente la fovea.
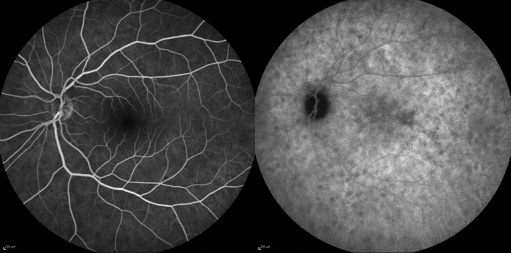
Procediamo nel nostro work up diagnostico sottoponendo la paziente all’indagine angiografica. L’esame con fluoresceina sodica ci permette di escludere la presenza di vasculite ed infiammazione del nervo ottico in quanto risulta sostanzialmente nella norma. L’angiografia con verde di indocianina mostra invece nelle fasi precoci la presenza di aree ipofluorescenti al polo posteriore che nei tempi tardivi diventano meno evidenti. Procediamo quindi richiedendo alcuni esami sistemici per escludere cause infettive ed infiammatorie di interessamento coroideale.
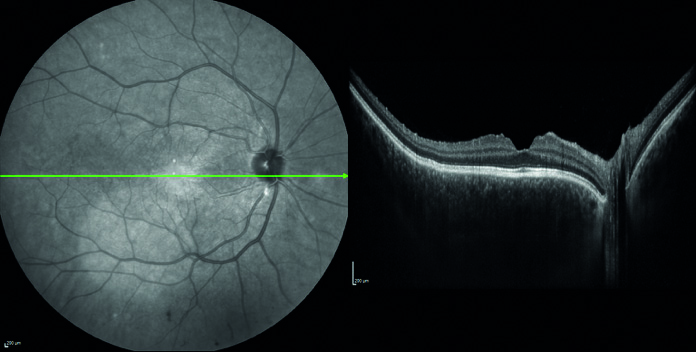
Emocromo, funzionalità epatica e renale, indici sistemici di infiammazione (VES e PCR), Quantiferon TB, anticorpi anti HBV, HCV, HIV e LUE risultano nella norma. La risonanza magnetica cerebrale conferma la presenza di un ispessimento della tonaca media oculare come già evidenziato dal nostro esame tomografico e la visita neurologica non rileva elementi patologici. Al successivo controllo dopo circa 7 giorni la paziente riferisce un soggettivo miglioramento della cefalea e del disturbo visivo, ma all’esame tomografico riscontriamo da una parte la riduzione del distacco del neuroepitelio perifoveale in occhio sinistro, dall’altra la comparsa di una nuova falda di fluido sottoretinico peripapillare nell’occhio destro.
Questo nuovo elemento rafforza una delle nostre ipotesi diagnostiche per confermare la quale eseguiamo, in collaborazione con i colleghi neurologici, una puntura lombare che evidenzia un quadro di pleiocitosi linfocitaria.



Diagnosi
Siamo quindi in grado di porre diagnosi di sindrome di Vogt Koyanagi Harada in quanto i criteri diagnostici della patologia rivisti e stilati nel 2001 sono soddisfatti: la nostra paziente non riferisce storia di trauma oculare, né di pregressa chirurgia, mostra un coinvolgimento oculare bilaterale, presenta all’esame tomografico ispessimento coroideale diffuso e fluido sottoretinico ed infine la rachicentesi ci conferma la presenza di pleiocitosi linfocitaria.
Si procede ad impostare una terapia steroidea ad alte dosi che prevede inizialmente la somministrazione di un bolo endovena, seguito da prednisone 1mg/kg/die per os per 15 giorni, da ridurre successivamente nei mesi successivi. Già un mese dopo il primo controllo la coroide risulta meno spessa, il liquido sottoretinico quasi completamente riassorbito e all’esame angiografico quasi del tutto scomparsi i granulomi precedentemente visibili nelle fasi precoci dell’esame con verde d’indocianina.
Sindrome di Vogt Koyanagi Harada Ci troviamo di fronte ad un caso di Vogt Koyanagi Harada, una panuveite granulomatosa che rappresenta il 4% delle uveiti negli Stati Uniti e l’8% di quelle in Giappone e la cui causa non è stata chiaramente definita sebbene sia stata ipotizzata una compartecipazione di fattori ambientali (contatto con EBV e CMV), genetici (associazione con HLA DR1 e HLA DR4) ed autoimmunitari (azione di linfociti T e B diretti contro melanociti coroideali).
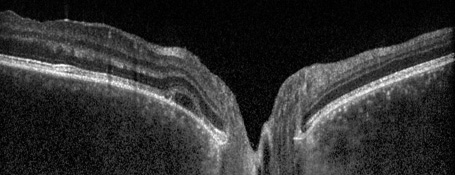

La patologia ha un andamento tipicamente cronico-ricorrente con una fase prodromica in cui si riscontrano sintomi sistemici quali nausea, febbre, cefalea, seguita da una fase di uveite acuta granulomatosa che interessa principalmente il segmento posteriore e causa calo visivo. Successivamente il paziente, soprattutto se non trattato tempestivamente, può andare incontro a un lungo periodo di convalescenza in cui compaiono i segni cronici della patologia: depigmentazione coroideale (Sunset glow) e limbare (Sugiura sign) con possibili esacerbazioni dell’infiammazione granulomatosa in camera anteriore e posteriore.
In ognuna di tali fasi possono essere presenti sintomi e segni di interessamento extraoculare a carico del distretto neurologico (meningismo, cefalea, ecc.), tegumentario (vitiligine, alopecia) ed uditivo (tinnito). La diagnosi differenziale va posta con cause infettive (infezione batterica, fungina, tubercolare o sifilitica, ecc.), infiammatorie (sarcoidosi, lupus, sclerite, ecc.) oncologiche (linfoma intraoculare o sistemico, ecc.) e traumatiche (oftalmia simpatica). Per identificare la patologia risulta fondamentale adottare un approccio multimodale mediante l’utilizzo di tomografia a coerenza ottica, auotofluorescenza, infrarosso e angiografia con fluoresceina e verde di indocianina.
Secondo la letteratura la puntura lombare non è necessaria per porre diagnosi, ma risulta di aiuto in casi di presentazione atipica o di forte sospetto diagnostico non avvalorato da altri elementi angiografici o sintomatici.
Nel caso della nostra paziente, per esempio, le caratteristiche angiografiche non risultavano fortemente indicative di VKH (assenza di hot disk, assenza di distacchi multipli del neuroepitelio con pooling di fluorescina), ma il riscontro di pleiocitosi linfocitaria alla rachicentesi ci ha permesso di porre diagnosi certa e quindi di iniziare tempestivamente la terapia steroidea. Impostare infatti una terapia immunosoppressiva ad alte dosi, precocemente e per lunghi periodi consente di ridurre le riacutizzazioni di malattia, accorciarne il decorso e ridurne le complicanze sul lungo periodo.





