


La retinoschisi X-linked (XLRS) è una patologia genetica che colpisce giovani individui di sesso maschile, caratterizzata da esordio precoce, tipicamente nella prima decade di vita, e prevalenza stimata tra 1/5.000 e 1/25.000.
L’alterazione anatomica tipica è una schisi retinica bilaterale degli strati nucleare esterno e nucleare intero, con coinvolgimento foveale quasi nel 100% dei casi. Rari casi con presentazione atipica possono fuorviare il clinico fino a formulare diagnosi errate.
Caso clinico
Afferisce presso il nostro ambulatorio di retina medica un uomo di 45 anni affetto da diabete mellito di tipo 1. Valutato precedentemente presso un altro centro, era stata formulata diagnosi di retinopatia diabetica non proliferante complicata da edema maculare meritevole di trattamento intravitreale con farmaci anti-VEGF. Null’altro da segnalare dal punto di vista anamnestico.
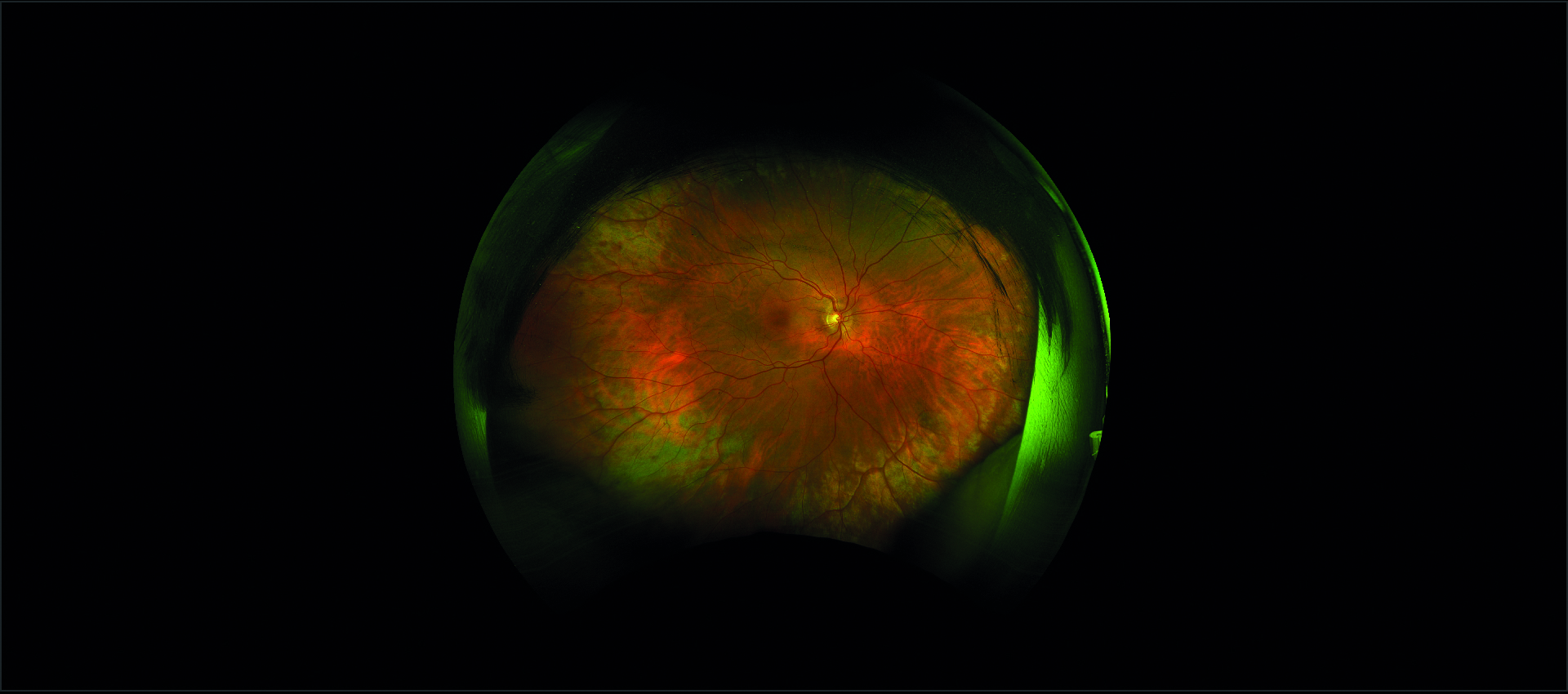
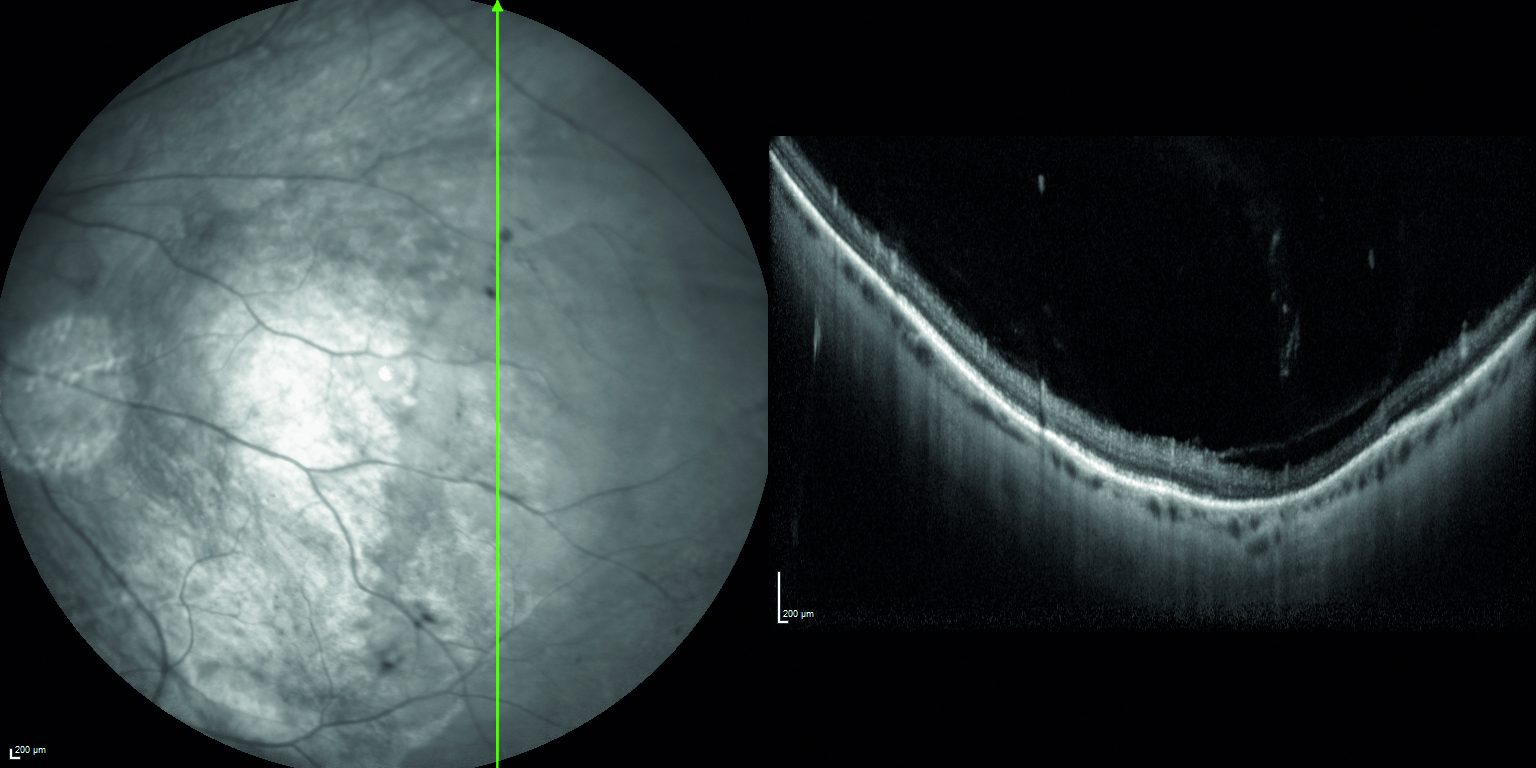
All’esame soggettivo della refrazione il paziente presenta un visus massimo di 8/10 in entrambi gli occhi e alla valutazione mediante lampada a fessura il segmento anteriore risulta essere nei limiti di norma, così come il tono oculare. All’esame del fundus oculi, eseguito in midriasi farmacologica, si riscontrano rari microaneurismi al polo posteriore ed un’alterata reflettività della retina periferica, quadro analogo in entrambi gli occhi (figura 1).
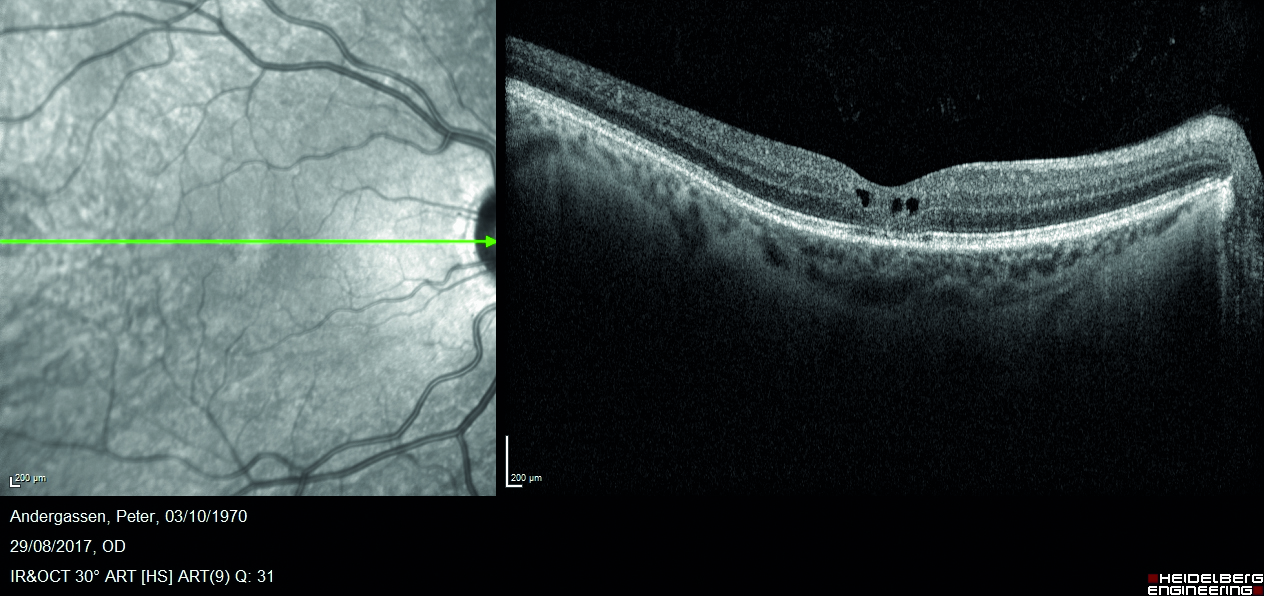
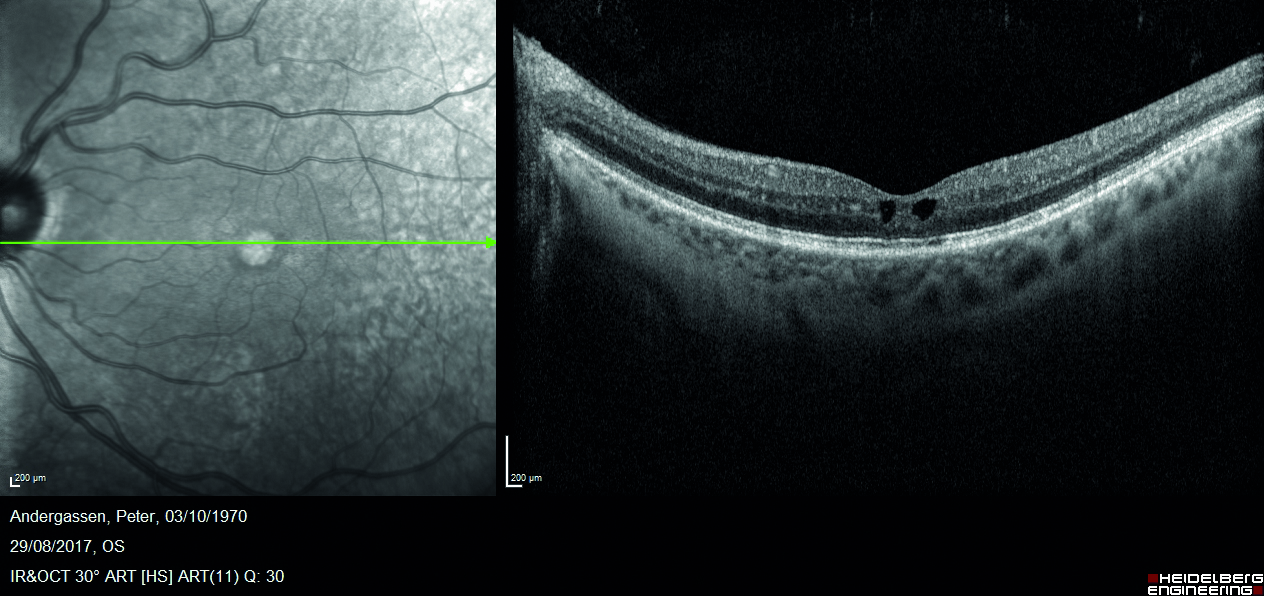
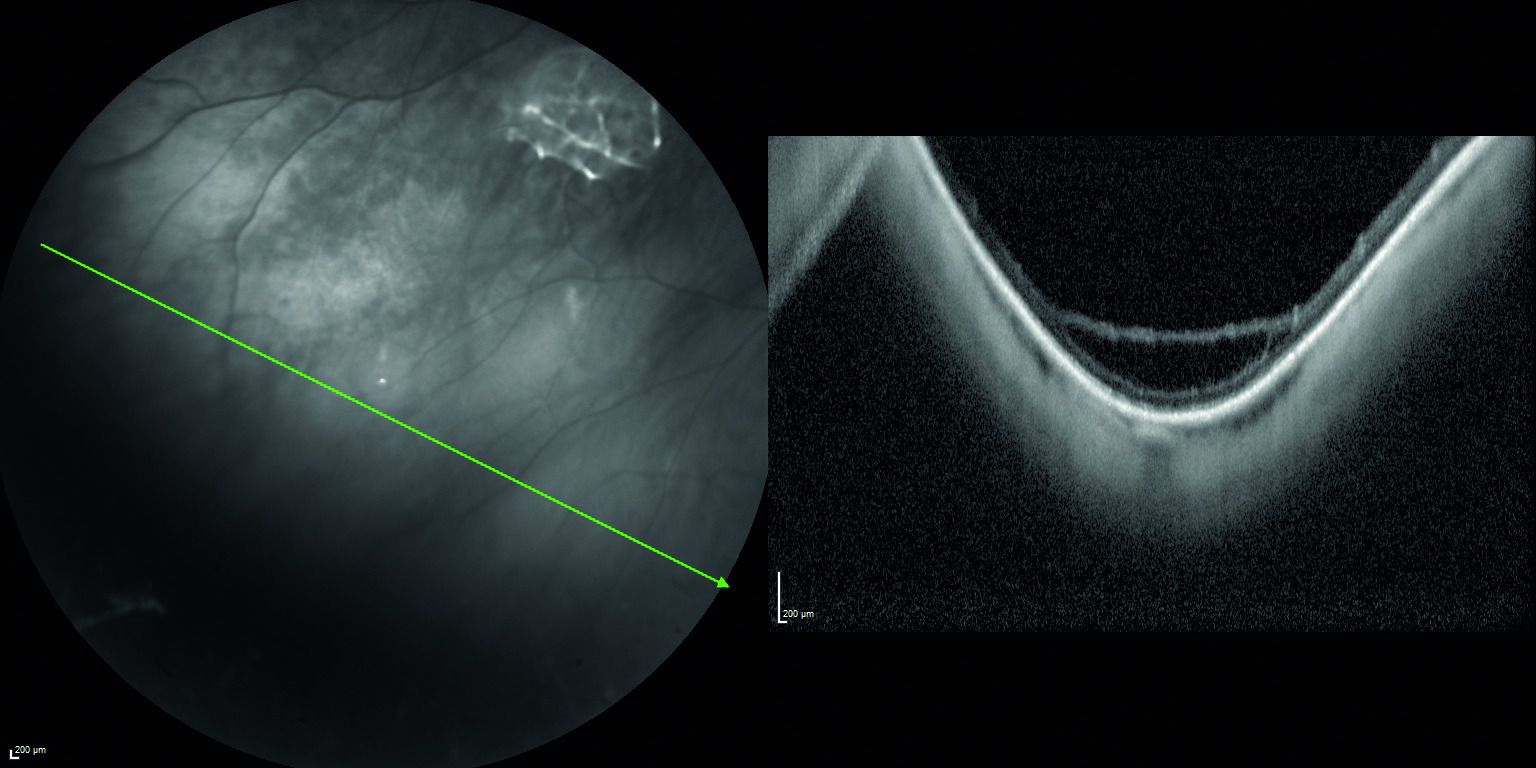
Mediante valutazione con tomografia a coerenza ottica (OCT) si riscontra la presenza di rare cisti intraretiniche in regione foveale, localizzate a livello degli strati retinici interni (figura 2) e schisi retiniche periferiche in corrispondenza delle aree di alterata reflettività (figura 3).
L’esame dell’autofluorescenza blu mostra come unici reperti patologici piccoli spot ipoautofluorescenti corrispondenti ai microaneurismi precedentemente descritti e aree di iperautofluorescenza in regione foveale dovute alla presenza delle suddette cisti intraretiniche (figura 4).
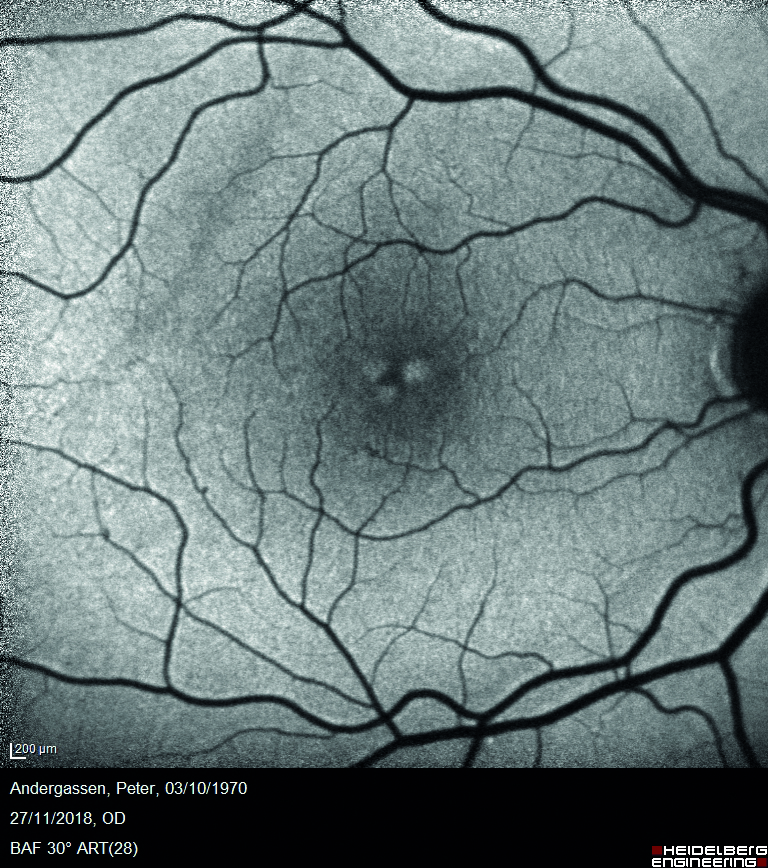

Quest’ultime dislocano gli strati retinici circostanti, tra cui lo strato plessiforme esterno contenente il pigmento maculare, e rendono quindi maggiormente visibile il segnale di autofluorescenza proveniente dall’epitelio pigmentato retinico.
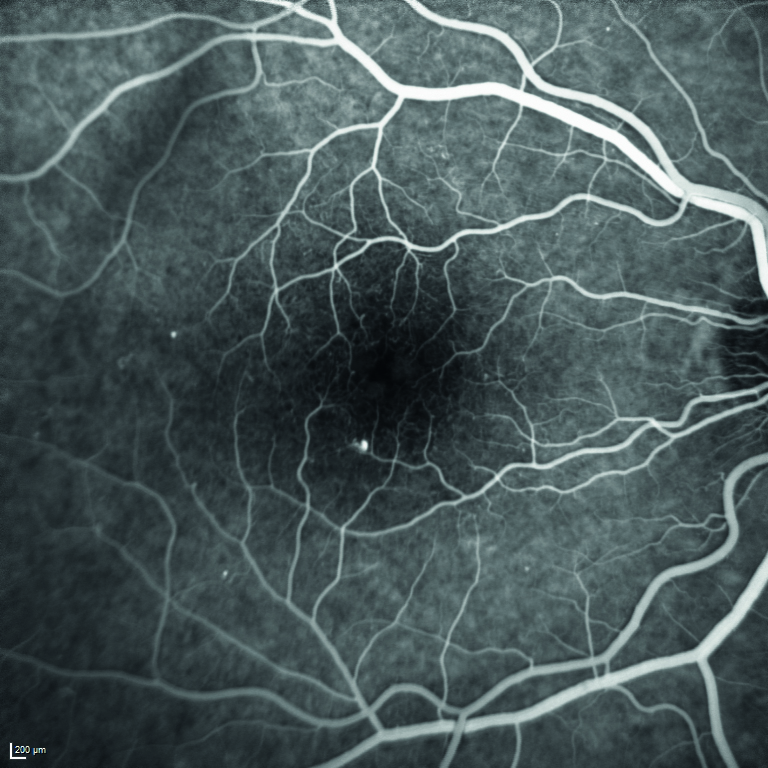
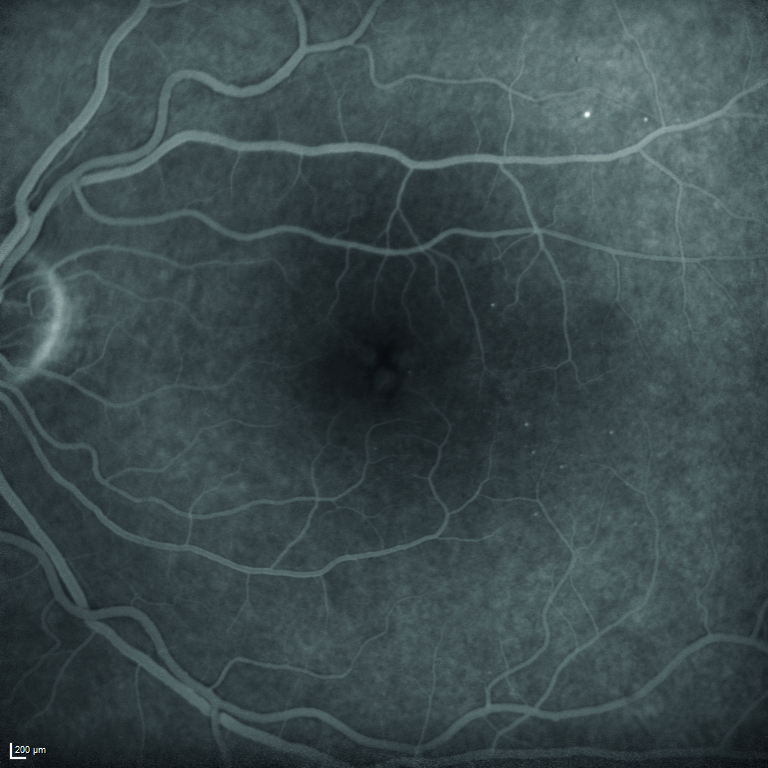
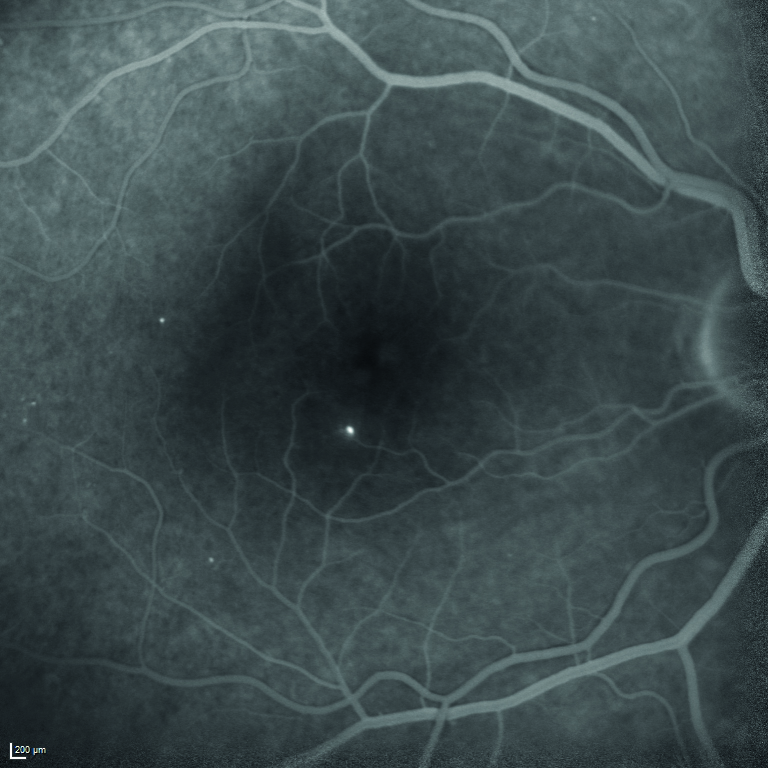
Viene quindi eseguita l’angiografia con fluoresceina sodica, ma il quadro maculare non risulta essere quello di un edema maculare diabetico per la mancata presenza di una franca alterazione della barriera emato-retinica nelle fasi tardive dell’esame (figura 5).

Alla luce della valutazione clinica e degli esami strumentali eseguiti, il sospetto diagnostico è di una patologia retinica genetica come la XLRS. Indagando ulteriormente l’anamnesi familiare del paziente, emerge che anche il nonno materno era affetto da disturbi visivi non meglio specificati, avvalorando l’ipotesi di una trasmissione matrilineare.
Eseguiamo quindi il test genetico che risulterà positivo per variazioni puntiformi e delezioni proprio del gene RS1, confermando quindi la diagnosi di XLRS.
Discussione
La retinoschisi X-linked può manifestarsi clinicamente con calo del visus e altri disturbi aspecifici come ipermetropia, strabismo e nistagmo. Il coinvolgimento è tipicamente bilaterale con progressione asimmetrica. L’esame elettroretinografico mostra una riduzione dell’ampiezza dell’onda b con onda a inizialmente conservata. Oltre alla caratteristica schisi foveale, si possono riscontrare strie radiali a livello della membrana limitante interna e schisi retiniche periferiche, presenti in almeno il 50% dei pazienti con prevalente localizzazione infero-temporale. Sono descritte anche anomalie microvascolari e, raramente, nevovasi retinici. Queste alterazioni strutturali predispongono, fin dalla giovane età, alla comparsa di emorragie vitreali e di distacchi di retina, il cui rischio aumenta poi con il passare del tempo.
A partire dalla quinta-sesta decade si assiste tipicamente ad un progressivo declino dell’acuità visiva; all’età di 60 anni l’acuita visiva media arriva ad essere 1/20, che viene considerato il limite nella definizione di cecità legale.
La retinoschisina, codificata dal gene RS1 ed espressa a livello dei fotorecettori e delle cellule bipolari, sembra infatti implicata nelle interazioni intercellulari e quindi nel mantenimento dell’adesione degli strati retinici tra loro. Più di 200 mutazioni del gene RS1 sono associate a questa patologia e la sua localizzazione sul braccio corto del cromosoma X ci spiega il coinvolgimento quasi esclusivamente del sesso maschile.
L’approccio terapeutico per questi pazienti è rivolto in primis alla correzione dell’errore refrattivo e dell’eventuale strabismo. Inoltre, in letteratura sono descritti casi in cui una terapia topica con inibitori dell’anidrasi carbonica ha determinato la riduzione del volume delle cisti e quindi migliorato l’acuità visiva. Tuttavia il loro impiego resta ancora controverso.
Prognosi della RSX
Nella RSXL la vista si reduce lentamente fino all'adolescenza, mentre nell'età adulta rimane relativamente stabile nella maggior parte dei pazienti. La malattia non progredisce fino ai 40 o 50 anni, età in cui insorge tipicamente un declino significativo dell'acuità visiva.
Attualmente grande interesse è rivolto alla terapia genica.
Modelli murini knock-out per il gene RS1 sono stati trattati con iniezioni intravitreali di un vettore virale (Adenovirus tipo 8) contenente il gene codificante la retinoschisina unito al suo promotore. Il risultato è un ripristino dell’espressione della proteina con conseguente miglioramento sia anatomico che funzionale.
Questa opzione terapeutica è stata recentemente studiata anche su pazienti affetti da XLRS e i risultati sono incoraggianti sotto il profilo della sicurezza e della tollerabilità, sia locale che sistemica. Inoltre, il riscontro in alcuni pazienti della riduzione degli spazi cistici, con conseguente miglioramento funzionale, suggerisce la possibilità di un futuro ingresso della terapia genica nel management di questi pazienti.
La genetica della RSX
La RSXL è ereditata come carattere legato all'X, perciò una femmina portatrice ha una probabilità del 50% di trasmettere la mutazione ai figli. Se la mutazione viene identificata in un membro affetto della famiglia, è possibile eseguire il test per identificare le femmine a rischio ed è possibile eseguire la diagnosi prenatale nelle gravidanze a rischio.





